Understanding FDA Classifications for Medical Devices
Navigating the FDA regulatory landscape can be daunting, especially for those new to medical device development. One of the foundational steps in this journey is understanding how the FDA classifies medical devices. Proper classification is crucial as it determines the regulatory requirements your device must meet. In this post, we’ll explore the FDA classifications, provide examples of each, and explain why knowing your device’s classification is essential for a smooth regulatory process.
What Are FDA Classifications?
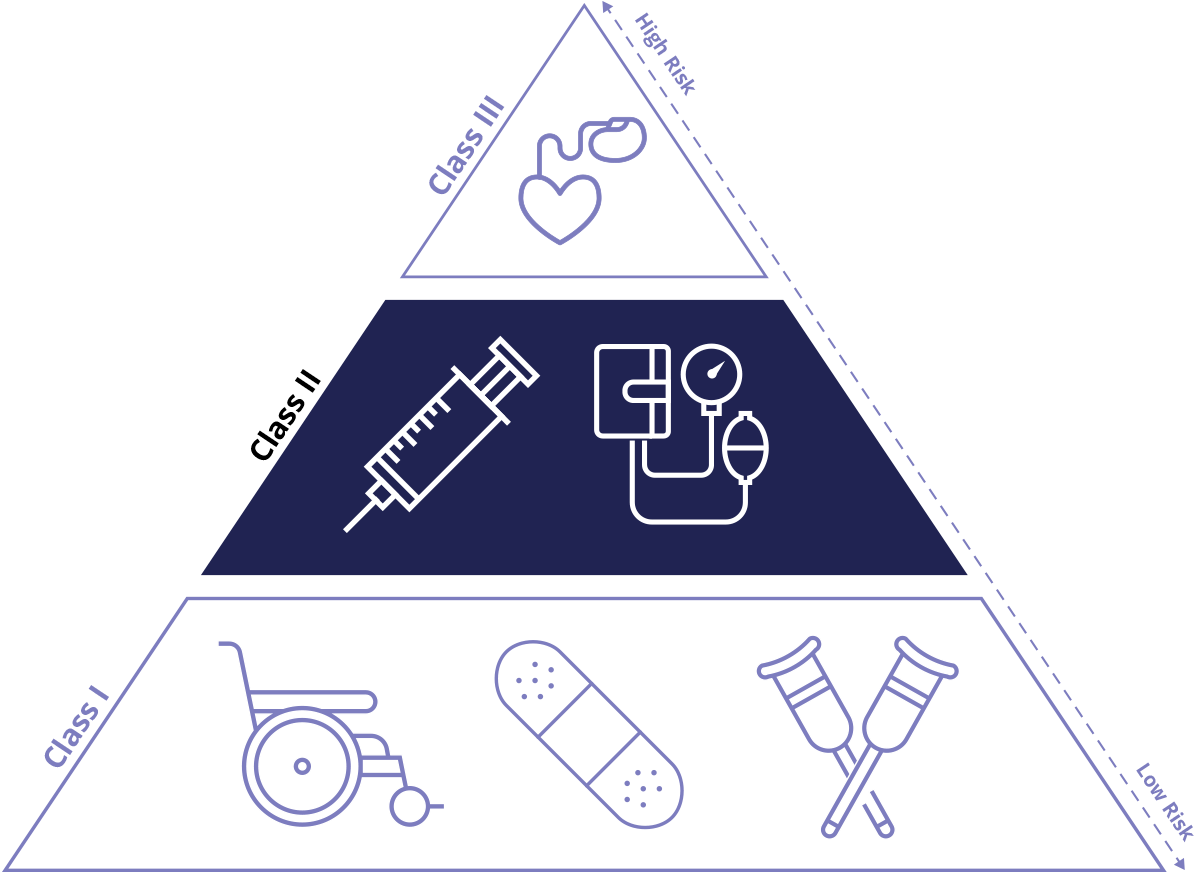
The FDA categorizes medical devices into three classes; Class I, Class II, and Class III. These classifications help determine the level of regulatory control necessary to ensure safety and effectiveness. Device classification is based on two distinct elements of the device, the intended use and the indications for use.
- Intended Use: Defined as the general purpose of the device or its function, and encompasses the indications for use.
- Indications for Use: Defined in 21 CFR 814.20(b)(3)(i) as the disease or condition the device will diagnose, treat, prevent, cure or mitigate, including a description of the patient population for which the device is intended.
In addition, the risk the device may pose to patients and users is a critical factor in determining its classification. Class I captures devices with the lowest risk profile, where as Class III includes those with the greatest.
Class I: Low Risk
Class I devices pose the lowest risk and are subject to the least regulatory control. Most Class I devices are exempt from premarket notification requirements, and General Controls are typically sufficient to ensure safety and effectiveness. However, you should perform a search of applicable regulations and ensure your device is exempt.
- Examples: Bandages, handheld surgical instruments, or examination gloves.
- Regulatory Requirements: General controls such as good manufacturing practices (GMP), labeling, and record-keeping.
Class II: Moderate Risk
Class II devices pose a moderate risk and require greater regulatory controls to provide reasonable assurance of their safety and effectiveness. These devices typically require premarket notification through the 510(k) process.
- Examples: Blood pressure cuffs, orthopedic implants, infusion pumps, or surgical drapes.
- Regulatory Requirements: General controls plus special controls like performance standards, post-market surveillance, and FDA guidelines.

Class III: High Risk
Class III devices are high risk and typically support or sustain life, are implanted, or present a potential unreasonable risk of illness or injury. These devices require pre-market approval (PMA) to ensure their safety and effectiveness.
- Examples: Pacemakers, heart valves, or implantable defibrillators.
- Regulatory Requirements: General controls and PMA, which includes a rigorous review of clinical data to support the device’s safety and effectiveness.
Why Is Classification Important?
Correctly classifying your medical device is essential because it dictates the regulatory pathway you must follow. Misclassification can lead to significant delays, increased costs, and potential non-compliance issues. Understanding the classification increases the chance of success in three main areas.
- Streamlines the Regulatory Process: Knowing your device’s classification helps you prepare the necessary documentation and follow the appropriate regulatory pathway from the start.
- Reduces Costs and Time to Market: Proper classification prevents unnecessary delays and expenses associated with incorrect submissions or additional testing.
- Ensures Compliance: Compliance with FDA regulations protects your company from legal risks and ensures that your device is safe and effective for users.
How to Determine Classification
Determining the classification of a medical device is not always a straightforward process. Three tools may be used to determine this; the Device Classification Panel, the Product Classification database, and the 510(k) Premarket Notificaiton database. Two important elements to understand when conducting your search:
- Regulation Number: A 7-digit ID that identifies a specific regulation within the Code of Federal Regulations (CFR). This is intended to group together a variety of medical devices that have similar indications and intended uses.
- Product Code: A three letter code, ex: “MJQ”, that captures a subset of medical devices that have the same indications and intended uses.
An important note here is that a single regulation number may contain multiple product codes, but each product code is intended to only apply to a single type of device.
The Device Classification Panel provides the ability to search for a classification by different medical specialties. Devices are grouped together by their 7-digit regulation number and provided a generic name. When reviewing a particular classification number the FDA will provide an Identification, a Classification (I, II, or III), and potentially any specific standards that may be applicable to the regulation.
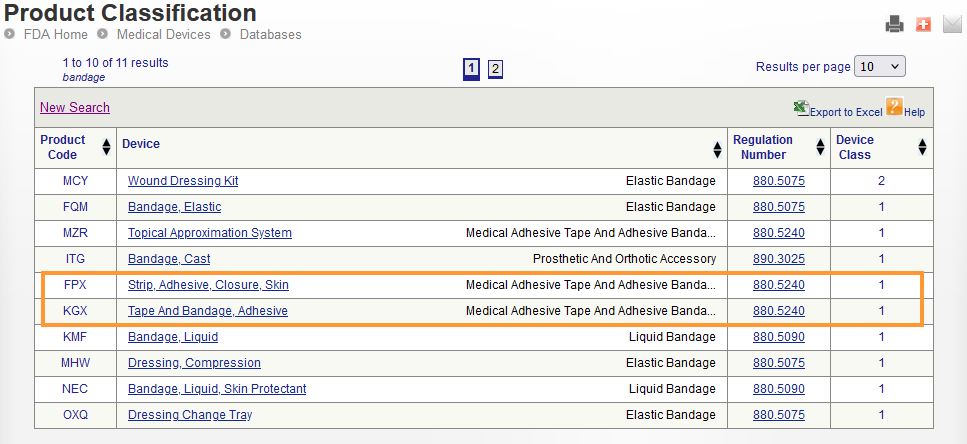
Searching the Product Classification database is generally more effective if you already have some known information about your device, or a similar device. For example, a submission type, a regulation number, the applicable review panel, or the device class. This database will provide a list of devices grouped by their product code. Reviewing a specific product classification will provide helpful information such as device class, regulation description, reporting requirements, and premarket review panel.
Premarket notifications are public information published by the FDA. If a predicate device exist on the market, and it was required to make a premarket submission, then you can locate it within this database. Using information like the product code, review panel, or device name will allow you to locate specific information about marketed devices. When reviewing a specific notification you can select “Summary” to access the notification that was submitted, usually containing helpful and relevant information about the product’s classification.
Using each of these tools can take some practice, and it is typically useful to use them in combination to best determine the applicable strategy for your product.
Summary
Understanding FDA classifications is a critical step in the medical device development process. By knowing the classification of your device, you can streamline your regulatory pathway, reduce costs, and ensure compliance. At Engineer Aid, we are committed to guiding you through every step of this process, leveraging our expertise to help you succeed in the competitive medical device market.
If you have questions about FDA classifications, or need assistance with your medical device development, contact us today for a consultation.